
Adults and children with congenital disorders of phenylalanine metabolism (PKU, HPA, BH4 deficiency) have been treated in the specialised PKU consultation for over 30 years.
Phenylketonuria (PKU) is a rare but one of the most common congenital metabolic disorders (1:8,000). It is inherited via chromosome 12.
Due to the lack of function of the enzyme phenylalanine hydroxylase (PAH), individual components of proteins that are ingested with food and produced in the body are only broken down incompletely. The amino acid phenylalanine cannot be broken down and phenylalanine accumulates in the body. If left untreated, this leads to mental developmental disorders, disability and epilepsy as early as infancy. With early, consistent treatment, normal development is the rule and the prognosis is good.
In most cases, the cause is a disorder of phenylalanine hydroxylase, in some cases there is a disorder of tetrahydrobiopterin metabolism (BH4), which has various influences on treatment. Men and women are equally affected.
A special situation arises in pregnant women with phenylketonuria, where the limit values must be adhered to particularly strictly in order to prevent damage to the foetus.
Contact person in the outpatient clinic
Ursula Strittmatter (dietary and nutritional advice)
Sandra Manz (paediatric nurse, centre coordination)
Make an appointment
Phone: 0731-500 57 292
Fax: 0731-500 57 298
Dietary/nutritional counselling
Medical treatment of concomitant diseases
Determination of phenylalanine levels (postal deliveries are also possible)
Advice on taking amino acid mixtures
Support during pregnancy with PKU
Drug treatment with BH4(Kuvan®) and pegvaliase (Palynziq®)
Scientific work to improve medical care for patients with PKU
Nowadays, the diagnosis is usually made shortly after birth via newborn screening. We advise and support families with a positive screening result on the path to the correct diagnosis and treatment.
At our centre, we can quickly initiate a comprehensive diagnostic programme (phenylalanine determination, biochemical analysis of BH4 synthesis disorders, genetic analysis) in order to subsequently discuss and implement possible treatment options.
Thanks to the analysis in our in-house amino acid laboratory, we can also react promptly to individual life situations through short informal channels.
With many years of expertise in diet and nutritional counselling, we are available for many questions and problems that arise in everyday life. We are in close contact and dialogue with suppliers of all common amino acid mixtures required as part of a diet and can provide you with professional advice on your choice.
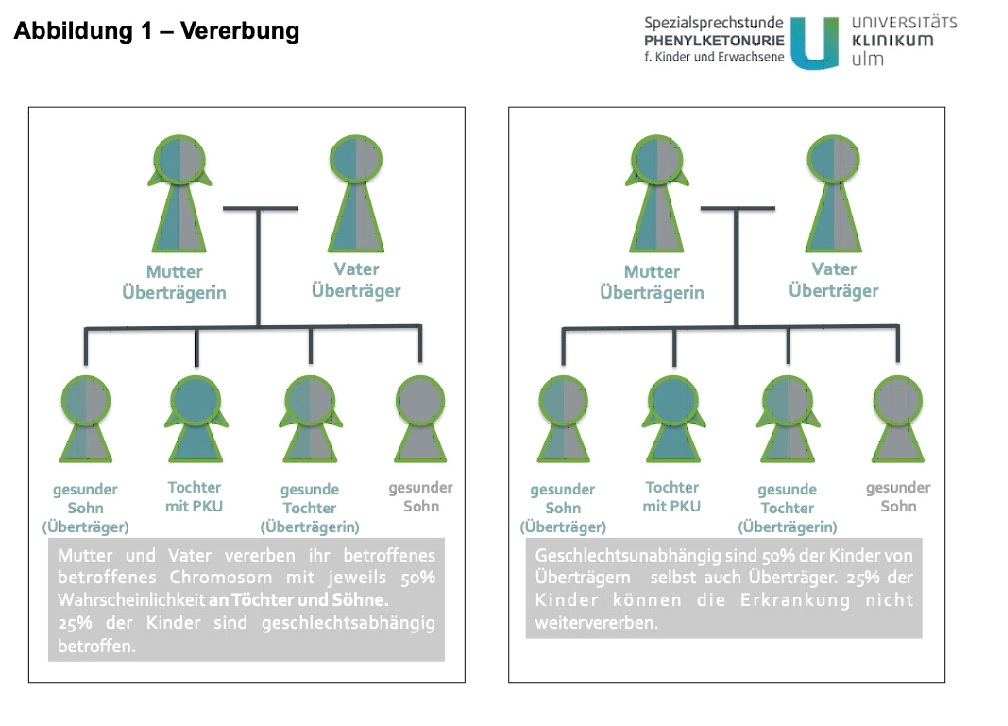
Phenylketonuria (PKU) is a hereditary disease. Every cell in the body contains 46 chromosomes (23 from each parent) on which the genetic information is stored. Two of these are the sex chromosomes X and Y. Men have one X and one Y chromosome, women have two X chromosomes.
The gene altered in PKU is located on chromosome 12, which means that men and women are equally affected by the disease.
Patients with PKU are certain (100%) to pass on an affected chromosome to their children. Healthy parents of children with PKU are carriers and have a 50% probability of passing on the affected chromosome (to both sons and daughters). Accordingly, the risk of repetition for siblings of a PKU patient is 25% in this case (see Figure 1).
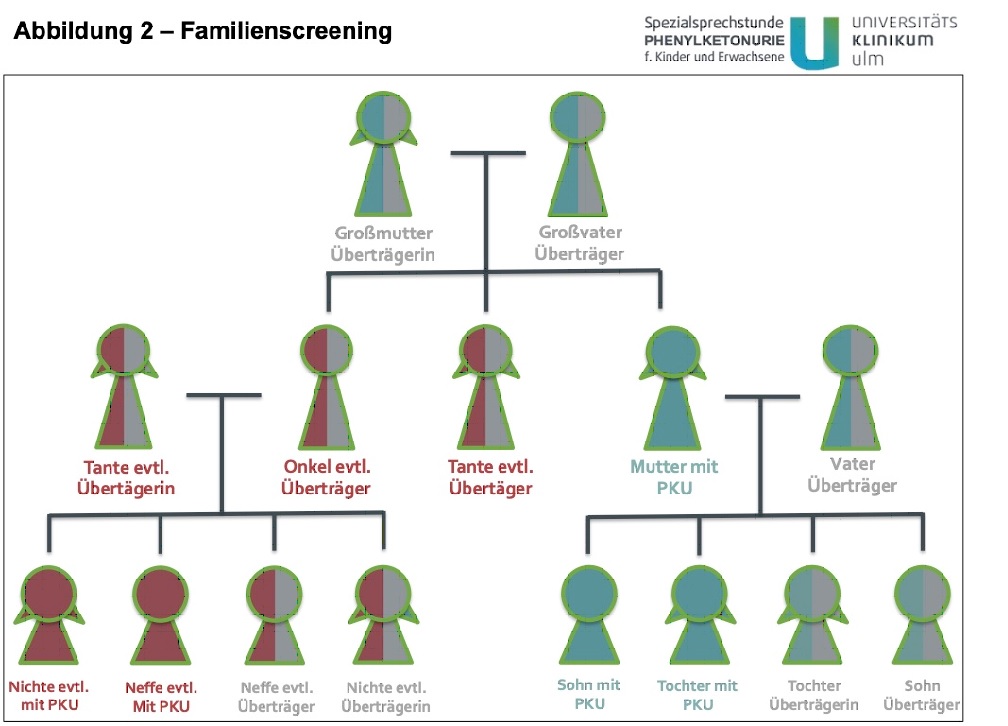
Affected children have inherited one affected chromosome from their mother and one from their father. Therefore, siblings of the affected person and siblings of the parents and grandparents of the affected person may also be affected or carriers of the disease. Accordingly, if a new case is detected, the relevant family members can be examined in order to assess the risk of recurrence for other family members. As part of a presentation in our specialist consultation, we can provide specialist human genetic counselling and identify any affected relatives and advise them if they wish to have children(qualification).