
In der Spezialsprechstunde PKU werden seit über 30 Jahren Erwachsene und Kinder mit angeborenen Erkrankungen des Phenylalaninstoffwechsels (PKU, HPA, BH4-Defizienz) behandelt.
Phenylketonurie (PKU) ist eine seltene, aber eine der häufigsten angeborenen Stoffwechselstörungen (1:8.000). Die Vererbung erfolgt über Chromosom 12.
Durch fehlende Funktion des Enzyms Phenylalaninhydroxylase (PAH) werden einzelne Bestandteile von Eiweißen, die über die Nahrung aufgenommen und im Körper produziert werden, nur unvollständig abgebaut. Die Aminosäure Phenylalanin kann nicht abgebaut werden und es kommt zu Anhäufung des Phenylalanins im Körper. Unbehandelt führt dies bereits im Säuglingsalter zu geistiger Entwicklungsstörung, Behinderung und Epilepsie. Bei frühzeitiger, konsequenter Behandlung ist eine normale Entwicklung die Regel und die Prognose gut.
In den meisten Fällen ist eine Störung der Phenylalaninhydroxylase ursächlich, in manchen Fällen liegt eine Störung des Tetrahydrobiopterinstoffwechsels (BH4) vor, was verschiedene Einflüsse auf die Behandlung hat. Männer und Frauen sind jeweils gleich schwer betroffen.
Eine besondere Situation ergibt sich bei Schwangeren mit Phenylketonurie, hier müssen die Grenzwerte besonders streng eingehalten werden um eine Schädigung des Fötus zu verhindern.
Ärztlicher Ansprechpartner
Ansprechpartner in der Ambulanz
Terminvereinbarung
Terminvereinbarung über das SPZ - Kontaktdaten siehe dort.
Diät-/Ernährungsberatung
Medizinische Behandlung von Begleiterkrankungen
Bestimmung des Phenylalaninspiegels (auch Posteinsendungen sind möglich)
Beratung bzgl. Einnahme von Aminosäurenmischungen
Unterstützung bei Schwangerschaft mit PKU
Medikamentöse Behandlung mit BH4(Kuvan®) und Pegvaliase (Palynziq®)
Wissenschaftliche Arbeit zur Verbesserung der medizinischen Versorgung von Patienten mit PKU
Heutzutage wird die Diagnose meist bereits kurz nach der Geburt über das Neugeborenenscreening gestellt. Wir beraten und begleiten Familien mit einem positiven Screeningbefund auf dem Weg zur richtigen Diagnose und Behandlung.
In unserem Zentrum können wir in kurzer Zeit ein umfassendes Diagnostikprogramm (Phenylalaninbestimmung, biochemische Analyse von BH4-Synthesestörungen, genetische Analyse) einleiten, um im Anschluss mögliche Therapieoptionen zu besprechen und durchzuführen.
Durch die Analytik in unserem hausinternen Aminosäurenlabor können wir durch kurze informelle Wege auch zeitnah auf individuelle Lebenssituationen reagieren.
Mit langjähriger Expertise unserer Diät- und Ernährungsberatung stehen wir für viele im Alltag auftauchende Fragen und Probleme zu Verfügung. Dabei stehen wir in engem Kontakt und Austausch zu Anbietern aller gängigen, von im Rahmen einer Diät notwendigen Aminosäuremischungen, und können Sie hier professionell bei der Auswahl beraten.
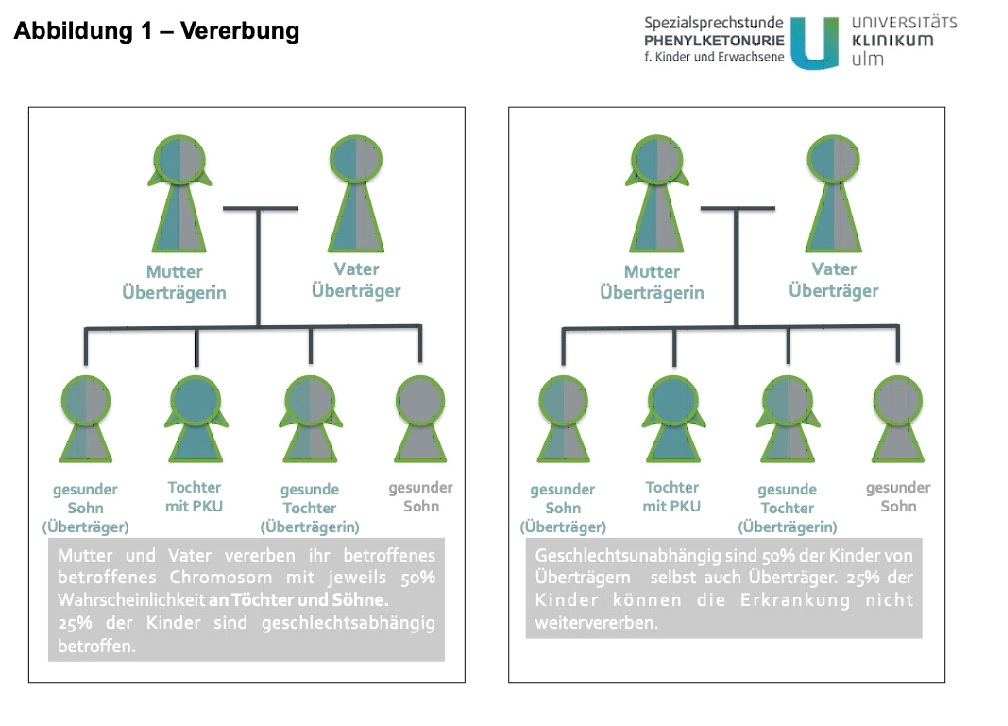
Phenylketonurie (PKU) ist eine Erbkrankheit. Jede Zelle des Körpers enthält 46 Chromosomen (23 von jedem Elternteil), auf welchen die Erbinformationen gespeichert sind. Zwei davon sind die Geschlechtschromosomen X und Y. Männer haben ein X- und ein Y-Chromosom, Frauen haben zwei X-Chromosomen.
Das bei PKU veränderte Gen befindet sich auf Chromosom 12. Das bedeutet, dass Männer und Frauen von der Erkrankung gleich schwer betroffen sind.
Patienten mit PKU vererben mit Sicherheit (100%) ein betroffenes Chromosom an ihre Kinder. Gesunde Eltern von Kindern mit PKU sind Überträger, und vererben mit einer Wahrscheinlichkeit von 50% das betroffene Chromosom (an Söhne wie Töchter). Entsprechend beträgt das Wiederholungsrisiko für Geschwisterkinder eines PKU-Patienten in diesem Fall 25% (siehe Abbildung 1).
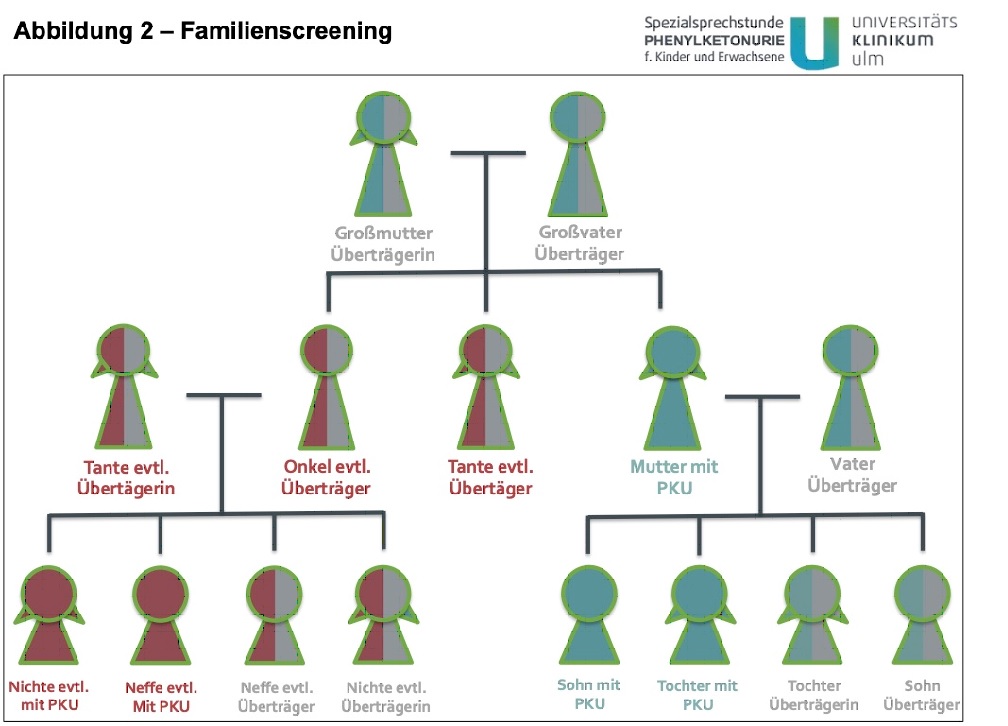
Erkrankte Kinder haben jeweils ein betroffenes Chromosom von der Mutter, und eines vom Vater geerbt. Daher sind möglichweise auch Geschwister der/des Betroffenen und Geschwister der Eltern und der Großeltern der/des Betroffenen erkrankt bzw. Überträger der Erkrankung. Entsprechend können bei Entdeckung einer Neuerkrankung die entsprechenden Familienmitglieder untersucht werden um das Wiederholungsrisiko für andere Familienmitglieder abschätzen zu können. Im Rahmen einer Vorstellung in unserer Spezialsprechstunde können wir eine fachgebundene humangenetische Beratung durchführen und möglicherweise betroffene Verwandte identifizieren und bei bestehendem Kinderwunsch beraten (Qualifikation).