Protein Aggregation and Prion-like Propagation
Toxic protein aggregation plays a significant role in the development and progression of various neurodegenerative diseases. In these conditions, specific proteins misfold and aggregate, leading to the formation of toxic protein aggregates that disrupt cellular functions and contribute to neuronal damage and cell death.
Besides intracellular protein aggregation, prion-like spreading of disease related proteins or aggregates has been observed in several neurodegenerative diseases. Protein aggregates, e.g. alpha synuclein (a-syn), propagate and spread between cells, contributing to the progression of the disease.
Aging is a significant risk factor for the development of neurodegenerative diseases. As individuals age, their cells and tissues undergo various changes, including a decline in cellular repair mechanisms, increased vulnerability to oxidative stress, and changes in protein homeostasis. These age-related processes can contribute to the accumulation or aggregation of proteins and cellular dysfunction, making the brain more susceptible to neurodegenerative conditions. Within the CRC 1506 “Aging at Interfaces” we want to understand whether cellular aging with all its associated altered mechanisms is the prerequisite for protein aggregation in general or whether the exposure time to misfolded proteins determines the accumulation of protein aggregates with their detrimental consequences. Further we want to understand whether pathological age-related mechanisms can be rescued by manipulation of structural cellular elements.
Parkinson’s disease (PD) is the second most common neurodegenerative disease with age being the strongest risk factor. Aggregation and oligomerization of alpha synuclein (a-syn) play a central role in the development of PD. Recently, we identified an age dependent accumulation of a-syn oligomers accompanied by neuronal cell loss and diminished motoric abilities using a PD in vivo model based on human a-syn protein complementation expressing α-synuclein split Gaussia luciferase (called S1/S2 model). This model allows highly sensitive assessment of a-syn oligomers since it is based on a human a-syn protein complementation assay that expresses a-syn split Gaussia luciferase. This mouse model is characterized by accumulation of a-syn oligomers accompanied by neuronal cell loss and diminished motoric abilities, as well as trans-cellular spreading of a-syn oligomers.
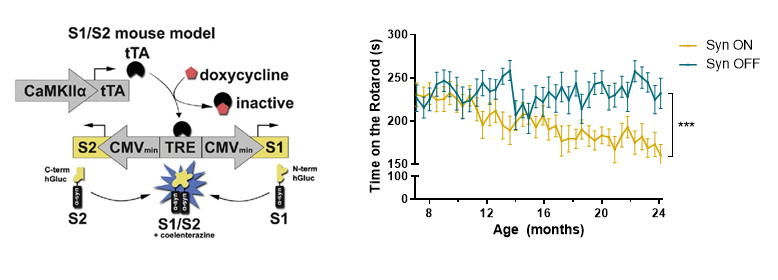
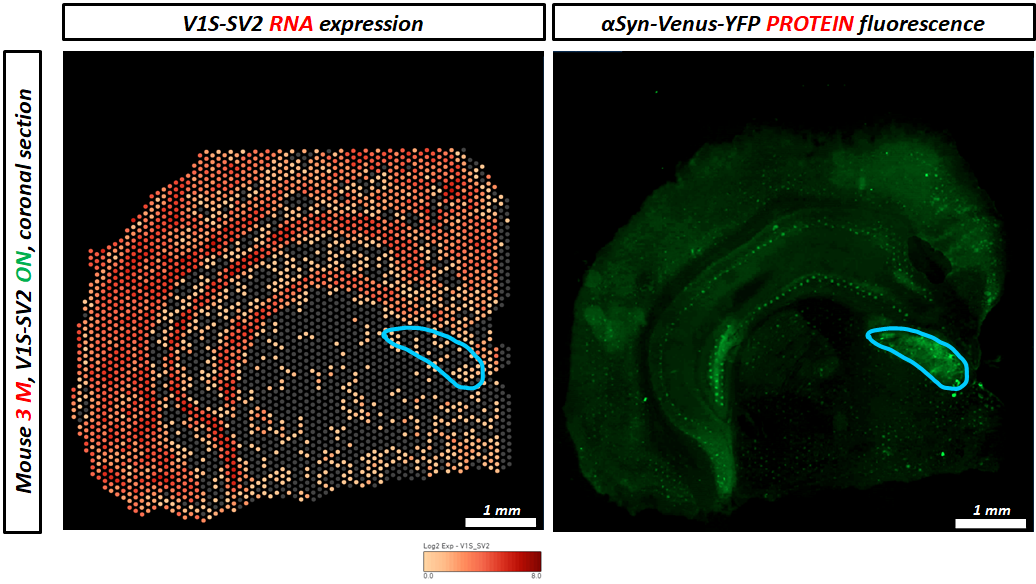
The underlying mechanisms responsible for an accumulation of a-syn oligomers during aging are not clear to date. The aim of this project is to understand whether neurons at a certain age are most vulnerable for a-syn oligomerization or whether the exposure time to those oligomers is responsible for the detrimental effects on neurons. We therefore induce a-syn oligomer expression for four months at different ages in our S1/S2 model and compare them with a-syn oligomer expression from birth regarding their motoric abilities. To determine whether motoric alterations correlate with a-syn oligomer load, we characterize a-syn oligomer species with biochemical methods. To get further insights into the underlying mechanism we are studying protein degradation mechanisms (autophagy and proteasomal activity), neurodegeneration, DNA damage, translational accuracy as well as perform single cell transcriptomics. Since we observed previously that a-syn oligomers can spread from neurons expressing a-syn to other neurons not expressing a-syn we want to determine whether cell to cell spreading is an age-related mechanism. To study cell-to-cell transmission on a single cell resolution we employ visium spatial transcriptomics and further investigate the consequences of a-syn spreading.
During aging, the activity of protein Cell division cycle 42 (Cdc42) increases in blood, liver and brain cells of the mouse. It has been shown that the increase of Cdc42 activity leads to disorganization in cells like hematopoietic stem cells and with this contributes to changes in age. Interestingly, a-syn oligomers can bind specifically to Cdc42 effector proteins. Recently, it has been shown that inhibition of Cdc42-activity by a specific inhibitor results ex and in vivo in a reorganization and rejuvenation of hematopoietic stem cells. Since Cdc42 activity is increasing with age, the question is whether modulation of Cdc42 activity might rescue the age-dependent Parkinson’s disease phenotype via changes on a-syn oligomerization. We are therefore pharmacologically target Cdc42 in PD in vivo models at different ages to test whether the PD phenotype and a-syn oligomers can be ameliorated by Cdc42 modulation.

Despite intensive research over the past decades, the interaction of trauma and its consequences on brain function and initiation of neurodegenerative diseases is not sufficiently unraveled. A link between traumatic injuries and their effects in the brain has been suggested, however, the exact cellular processes involved, as well as the mechanisms of how this happens are still mostly unknown. As trauma has been implicated in the development of Parkinson’s disease (PD) we aim to unravel within the CRC 1149 “Danger Response, Disturbance Factors and Regenerative Potential after Acute Trauma” the underlying mechanisms linking trauma and inflammation to neurodegeneration.
On the molecular level, brain injury has been suggested to facilitate a-synuclein (a-syn) aggregation, which is the key protein in PD and the main pathological hallmark in the PD brain. Increased levels of a-syn were reported in the cerebrospinal fluid of patients after TBI and a-syn immunoreactive neurons were seen in resected cortical tissue after a severe TBI in humans. Besides TBI also traumatic injuries and inflammation in the periphery increase the risk of developing neuropsychiatric and neurodegenerative diseases. However, our knowledge on the pathophysiology linking peripheral inflammation to brain malfunction is still limited. We could show that a peripheral injury like a thoracic trauma leads to a strong increase in a-syn oligomers while the total a-syn amount remained constant, suggesting that peripheral injury influences protein aggregation in the brain of PD models.
Together with the group of Leda Dimou we are aiming to understand to what extent a TBI or a sepsis would change the physiological function of the brain regarding gliosis, adult neurogenesis, protein aggregation, a-syn spreading and neurodegeneration. Additionally, we want to answer whether a preceding trauma can exacerbate or even trigger PD pathology on the long term, not only on a cellular level but also regarding behavioral changes. On the other hand, we are asking how a comorbidity such as PD can alter the outcome of a traumatic lesion at short timepoints after an injury.
In 10 % of ALS cases, the disease is inherited and caused by mutations in different genes such as C9ORF72,SOD-1 and TARDBP. TARDBP encodes the transactive response DNA-binding protein 43 kDa (TDP-43), which seems to play a critical role in ALS. 97 % of all ALS cases, independent of the familial background, show TDP-43 positive cytoplasmic inclusions. In ALS, TDP-43, normally predominantly located in the nucleus, mislocalizes in the cytoplasm and forms cytoplasmic TDP-43. Therefore, protein misfolding and aggregation seems to be a critical process in the cause of the disease. Heat shock protein 70 kDa (Hsp70), a molecular chaperon, has been identified to be neuroprotective in several neurodegenerative diseases. For ALS, it has been reported that Hsp70 regulates TDP-43 aggregation in the heat shock response and could prevent aggregation of the 25-kDa C-terminal fragment of TDP-43. The aim of this project is to study the effect of Hsp70 family members on TDP-43 aggregation, localization and solubility. We focus not only on Hsp70, but also on neuron specific Hsp proteins. To this end we employ TDP-43 cell culture models as well as patient cells and study TDP43 aggregation, localization as well as binding and interaction of TDP-43 and Hsp70 family members.
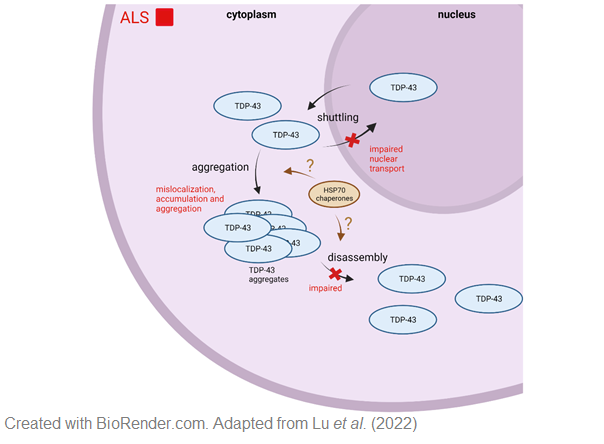
Since TDP-43 aggregation is the main neuropathological hallmark of ALS we aim to develop in vivo models that allow a quantitative and spatial resolution of TDP-43 aggregation.
To this end human WT TDP-43 is tagged with hGaussia luciferase or split venus YFP, called TDP-43-Luci or TDP-43-V1/V2 (TV1/TV2), respectively. TDP-43 is fused to either N- or C-terminal non fluorescent Venus YFP that reconstitute to fluorescent Venus VFP upon aggregation. To aim towards inducible and cell type specific TDP43 expression, we employ a Tamoxifen inducible Cre-ERT2 system.
With these models we will study the potential correlation between TDP-43 aggregation and an ALS phenotype, as well as in vivo TDP-43 aggregation and propagation in a quantitative as well as spatial resolution. We pay particular attention to TDP-43 transmission from neuronal-to-neuronal cells as well as also to non-neuronal cells and study its subsequent consequences.

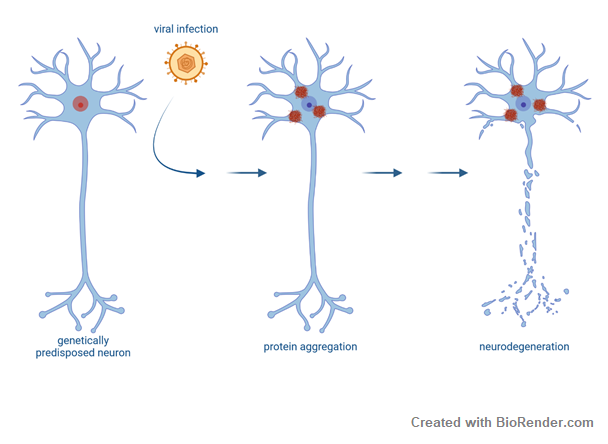
Development of ALS is likely caused in a multi-step process. External factors might act on top of a genetic predisposition to trigger disease onset. In this project we study whether viral infections and the subsequent antiviral immune response impact ALS pathogenesis. Viral infections might have either happened in early adulthood or later in life. Whether this is clinically and epidemiologically linked to ALS needs to be explored. On a molecular level, viral infections have recently been shown to induce stress granules and FUS proteinopathy in ALS models. Therefore, we also aim elucidate together with the group of Konstantin Sparrer the molecular mechanisms underlying viral infections on TDP43 aggregation as potential risk factors for development and/or progression of ALS.
People

Verena Bopp
M. Sc. Molecular and Translational Neuroscience
Phone: +49731-500-63039
Fax: +49 731-50063050
Mail: verena.bopp@uni-ulm.de

Alicia Goreth
M.Sc. molecular medicine
Phone: +49731-500-63061
Fax: +49 731-50063050
Mail: alicia.goreth@dzne.de

Dr. Sarah Tripke
M.Sc. Biochemistry
Phone: +49-731-500-63061
Fax: +49-731-500-63050
Mail: sarah.brockmann@uni-ulm.de

Daniel Rombach
M.Sc. Biochemistry
Phone: +49731-500-63061
Fax: +49 731-50063050
Mail: daniel.rombach@dzne.de

Dr. Eva Buck
M.Sc. Molecular Biosciences
Phone: +49731-500-63061
Fax: +49 731-500-63050
Mail: eva.buck@uni-ulm.de

Isabel Hagmann
Pharmazeutische Biotechnologie
Phone: +49-731-500-63043
Fax: +49-731-500-63060
Mail: isabel.hagmann@hochschule-bc.de

Jaehyun Lee
Phone: +49731-500-63152
Fax: +49 731-50063050
Mail: Jaehyun.Lee@dzne.de